- Introduction
- Medicinal products containing or consisting of GMOs
- Decision-making
-
Tools for risk assessment and risk management (opens in a new page)
-
Some figures (opens in a new page)
Introduction
Prior to marketing, all medicinal products derived from biotechnology (therefore including medicinal products containing or consisting of GMOs) must obtain an authorisation issued by the European Commission upon advice of the European Medicines Agency (EMA). Access to the Community market for GMO medicinal products derived from biotechnology is subject to the centralised procedure laid down in Regulation (EC) 726/2004. If authorisation is granted, it is automatically valid for all Member States of the European Union.
The applicant for a marketing authorisation for a biotechnological medicinal product shall submit a dossier for registration to the EMA that will be assessed on the basis of scientific criteria concerning the quality, safety and efficacy of the medicinal product concerned. According to the centralised procedure and depending on the type of medicinal product, the EMA's assessment of a medicinal product is performed either by the Committee for Medicinal Products for Human Use (CHMP), by the Committee for Medicinal Products for Veterinary Use (CVMP) or by the Committee for Advanced Therapies (CAT).
For each dossier the assessment is done under the coordination of a rapporteur and a co-rapporteur appointed by the CHMP, the CVMP or the CAT. During the procedure a draft assessment report is first prepared, on the basis of which questions can be sent to the applicant. Following the applicant's replies, a final assessment report is submitted for discussion at the relevant EMA commitee. Once the evaluation is completed the CHMP, CVMP or CAT discusses the risk versus the benefit of the use of the product and finalises its opinion. When positive, the opinion of EMA is transmitted to the Commission that will make the final decision.
Medicinal products containing or consisting of GMOs
Medicinal products containing or consisting of GMOs constitute a special regulatory case due to reciprocal provisions in the Directive 2001/18/EC (Article 12.2) and Regulation (EC) 726/2004 (Articles 6.2 and 6.3). In this case the application shall also include an environmental risk assessment (ERA) in accordance with the provisions of Annex II of Directive 2001/18/EC. It shall therefore contain the complete technical dossier supplying the information required by Annexes III and IV of Directive 2001/18/EC.
The ERA applies to medicinal products containing or consisting of GMOs, but not to medicines produced from GMOs. The latter include, for example, insulin produced from recombinant bacteria or, a more recent development, human anticoagulant protein present in the milk of genetically modified goats.
The ERA is reviewed in consultation with the bodies established by the Community or the Member States, in accordance with Directive 2001/18/EC. In Belgium, this risk assessment is evaluated by the Biosafety Advisory Council, with the scientific support of external experts and the SBB.
Inputs of the Biosafety Advisory Council and the SBB (see figure below)
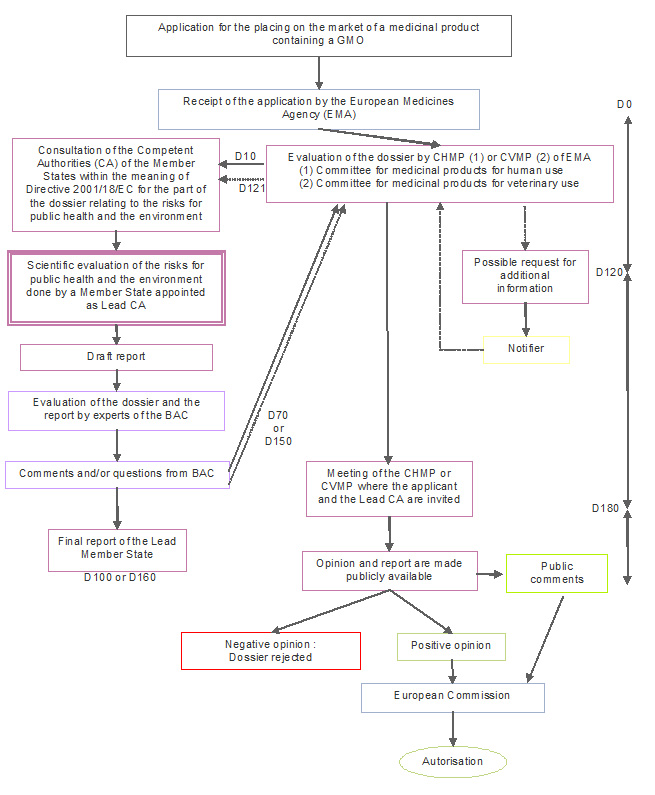
The draft assessment reports prepared by the EMA rapporteur and co-rapporteur and validated by the CHMP or the CVMP contain a section specifically dealing with the environmental risk assessment (ERA) of the GMO medicinal product. The ERA relates to the risks to the biotic environment, as well as to the potential hazards of the GMO for those in the vicinity of the person treated or for health care personnel, and to the risks to public health. In this section of his report, the rapporteur highlights any shortcomings in the ERA and, if necessary, proposes a list of questions to be sent to the applicant.
The ERA section of the report is communicated to the Biosafety Advisory Council (BAC) (and to all designated Competent Authority of the other member states), who has beforehand been granted access to the ERA part of the dossier.
The BAC drafts and send to the EMA its opinion on the report of the EMA rapporteur relating to the ERA, eventually pinpointing issues which the rapporteur may not have considered. The BAC's advice indicates any additional information that should be obtained to supplement the ERA. The BAC provides its opinion to EMA on each further assessment reports drafted by the rapporteur and co-rapporteur as a result of the applicant's replies.
Decision-making
As mentioned before, the European Commission takes the final decision to place on the market a medicinal product that is eligible for the centralised procedure.
The full scientific assessment reports of every medicine granted a central marketing authorisation by the European Commission following an assessment by the EMA's CHMP or CVMP are publicly available on the EMA website (EPAR - European Public Assessment Report). The EPAR includes the Product information leaflet and a Summary of the product characteristics in several European languages as well as the scientific discussion and the steps taken for assessment (in English).
Marketing authorisations are valid for five years. Applications for renewal must be made to the EMA at least six months before this five-year period expires.